PACKMOL is a free software for building initial configurations for Molecular Dynamics. This software combines optimization tools with physical-chemical insight and is the result of a partnership of the AOG with Prof. Leandro Martínez, from the Institute of Chemistry – Unicamp, who is in charge of its maintenance. The main paper that describes Packmol has 355 citations in Webofscience (Core Collection). Packmol has been downloaded 11547 times from 6932 different users.
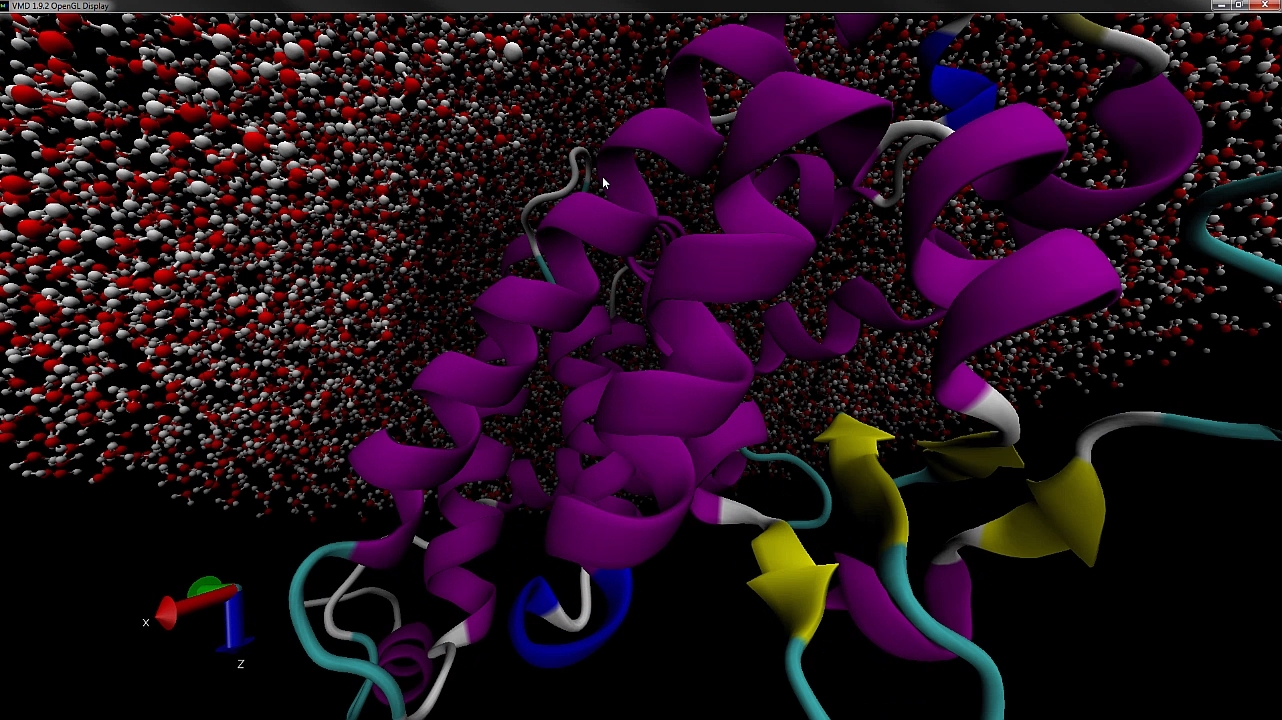
Packmol creates an initial point for molecular dynamics simulations by packing molecules in defined regions of space. The packing guarantees that short range repulsive interactions do not disrupt the simulations.
The great variety of types of spatial constraints that can be attributed to the molecules, or atoms within the molecules, makes it easy to create ordered systems, such as lamellar, spherical or tubular lipid layers.
The user must provide only the coordinates of one molecule of each type, the number of molecules of each type and the spatial constraints that each type of molecule must satisfy.
The package is compatible with input files of PDB, TINKER, XYZ and MOLDY formats.
L Martinez, R Andrade, E G Birgin and J M Martinez. PACKMOL: A package for building initial configurations for molecular dynamics simulations. Journal of Computational Chemistry 30(13):2157–2164, 2009. URL, DOI
Jose Mario Martinez and Leandro Martinez. Packing optimization for automated generation of complex system’s initial configurations for molecular dynamics and docking. Journal of Computational Chemistry 24(7):819–825, 2003. URL, DOI